下载或打开 医联APP 查看完整评论
立即下载
打开APP
间断呕血、腹痛病例?
主诉 病史
患者男性,31岁,主诉为“9个月间断呕血4次,腹部胀痛10d”。该患者曾因反复消化道出血入院,此次因腹胀明显就诊于吉林大学白求恩第一医院肝胆胰内科。曾行胃镜检查提示食管胃底静脉曲张、门静脉高压,腹部彩超提示“肝硬化、脾大、门静脉高压”,2013年2月行脾栓塞治疗(栓塞40%)。 既往:儿童时期曾诊断为“肠病性肢端皮炎”;颈、躯干部皮肤黑变14年余,渐加重,2012年8月行颈部皮肤活组织检查,病理诊断为“血管萎缩性皮肤异色病”,未正规治疗。其父亲有肝硬化病史。
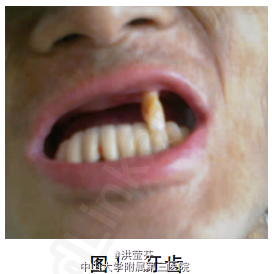
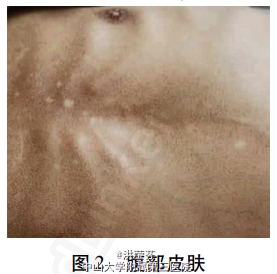
查体 辅查
入科查体:早衰面容,重度贫血貌,毛发稀疏,牙齿脱落(图1),巩膜呈淡蓝色,颈、胸腹部、躯干、四肢见皮肤弥漫对称性黑色斑片及斑疹(图2),未见肝掌、蜘蛛痣,指(趾)甲脱失;腹膨隆,腹壁静脉曲张明显,移动性浊音阳性,双下肢轻度水肿。 辅助检查:血常规提示重度贫血(血红蛋白原125(CA125)223.29U/ml、甲胎蛋白7.67ng/ ml、性激素检查提示孕酮及泌乳素升高而睾酮下降(经内分泌科会诊后排除其相关疾病);自身免疫性肝病相关抗体检测均为阴性;肝豆状核变性及血色病相关检查未见异常;腹水常规提示漏出液改变,红细胞1.42×1010/L;全腹CT平扫提示肝硬化,腹水、脾大,考虑门静脉高压伴侧支循环开放。 该患者的皮肤活组织病理检查结果为:棘层萎缩变薄,真皮浅中层血管周围中等量淋巴细胞浸润伴大量嗜色素细胞(图3)。

诊断 处理
入院后临床诊断为“肝硬化失代偿期、先天性血管萎缩性皮肤异色病”,给予常规保肝、利尿等治疗,患者病情好转后出院。目前处于院外定期随访中,病情相对稳定。
随访 讨论
先天性血管萎缩性皮肤异色病又称Rothmund-Thomson综合征(RTS),1868年由Rothmund首次描述,1957 年由Taylor提出。 本病属于一种罕见的常染色体隐性遗传性皮肤病,分为Ⅰ型RTS及Ⅱ型RTS。 目前Ⅰ型RTS致病基因尚不明确,Ⅱ型RTS致病基因为 RECQ14,它主要在婴儿期起病,主要表现为骨骼异常和恶性肿瘤。 研究发现有RECQ14基因突变的患者都伴有骨发育异常,预示该基因的突变可能与骨肉瘤及癌症的发生有关。 临床表现包括皮肤颜色改变、毛发稀疏、甲营养不良、牙釉质发育不全、缺牙或多生牙,68% 的患者出现骨骼发育异常。 它是一种罕见的蕈样真菌病的变种,其临床特点是广泛的网状或斑马纹状的过角化鳞片状丘疹。其病理改变为真皮基层轻度空泡变性,真皮浅层血管周围黑素颗粒及噬黑素细胞较多,血管轻度扩张、充血。患者自述儿童时期曾诊断为“肠病性肢端皮炎”。 文献记载,肠病肢端皮炎1943年由 Danbolt 和Closs 首次报道,又名 Danbolt-Closs综合征、Brandt综合征。该病是一种常染色体隐性遗传病,婴儿期发病,青春期好转,成人极少发病,有独特的三联症:肢端皮炎、秃发和腹泻,但三者常不同时出现。 肝硬化是各种慢性肝病发展的晚期阶段,病理上以肝脏弥漫性纤维化、再生结节和假小叶形成为特征,晚期以肝功能减退和门静脉高压为主要表现。 来源:医脉通http://case.medlive.cn/liver/case-article/show-79253_39.html
发布于 15-06-15 00:09