下载或打开 医联APP 查看完整评论
立即下载
打开APP
甲状腺激素受体基因突变一例
主诉 病史
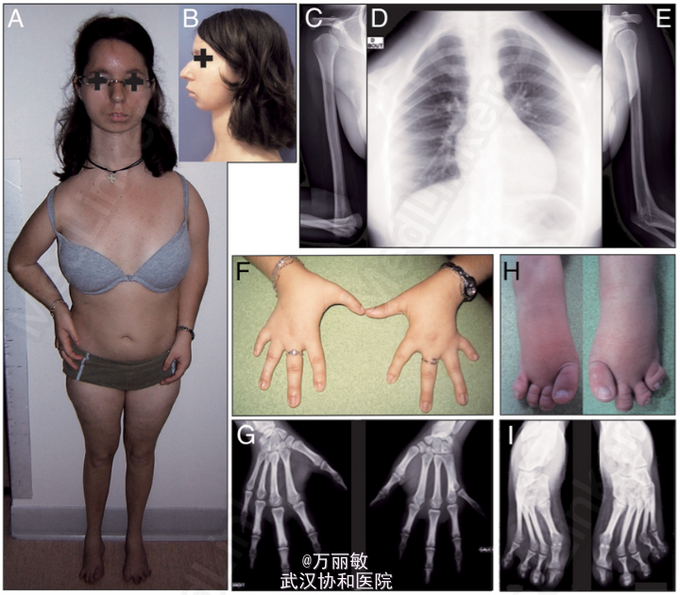
患者,女,25岁,因“生长发育迟缓”入院。 患者自幼生长发育均较同龄人迟缓,并伴有严重的骨发育不良,表现为锁骨发育不全、手指及足趾畸形及肘关节畸形等。12岁开始患者出现血钙升高,PTH水平升高,诊断为甲状旁腺机能亢进,后因出现反复高钙血症,复发性肾结石,手术切除部分甲状旁腺。现为求进一步诊治入院。既往史:先天性室间隔缺损(自行闭合),长期慢性腹泻。
查体 辅查
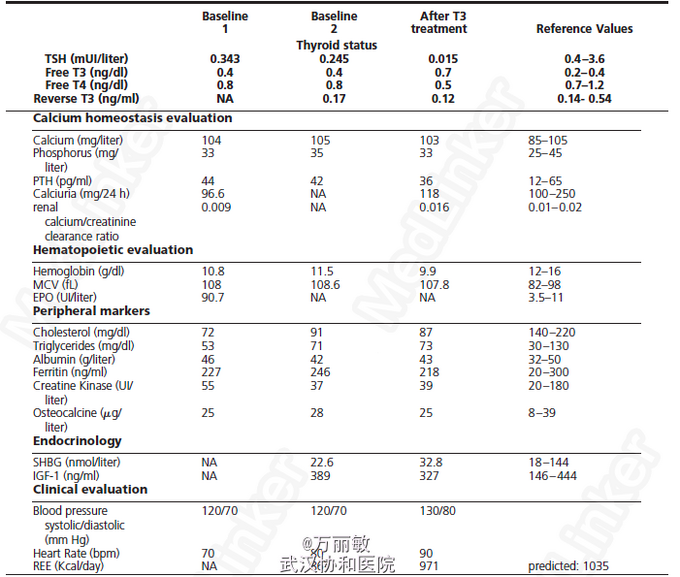
查体:图1 辅检:生化指标(图2),静息能量消耗(REE)降至预测值的80.5%;Wechsler成人智能量表显示患者智力处于平均偏下水平。骨髓电镜检查显示1型先天性红细胞发育不全。头颅MRI、泌乳素、皮质醇水平均正常。 与高钙血症相关的基因(CASR MEN1和HRPT2)、与先天性红细胞发育不全相关基因(CDAN1, SEC23B and KLF1)均未见突变。由于患者表现出一些过去曾报道的甲状腺激素受体TRα突变疾病(RTHα)的症状和体征(生长发育迟缓、巨头畸形、低基础代谢率、FT4/FT3比值降低,先天性贫血),故对编码TRα的基因THRA进行了测序,发现一个单等位基因突变(c.1075C>G),进而导致其编码蛋白中的一个氨基酸改变(N359Y)。替换的氨基酸位于TRα的配体结合域,导致该受体蛋白与T3亲和力降低。
诊断 处理
诊断考虑为THRA突变导致的甲状腺激素抵抗(RTHα) 给予碘塞罗宁(25ug/d)一个月后,患者T3水平上升,TSH及T4水平下降,基础代谢率升高,然而腹泻加重,心率增快,其他生化指标没有明显变化。
随访 讨论
RTHα非常罕见,过去仅有7例报道,且基因型几乎各不相同(2例TRα1E403X,1例TRα1F397fs406X,1例TRα1A382fs388X , 1例TRα1A263V, 1例TRα1C392X,1例TRα1P398R ),本病例为一个新的基因型TRα1N359Y。RTHα临床表现变化多端,其共同的临床表现有:生长发育迟缓、巨颅畸形、特殊的面部特征、FT4/FT3比值降低、先天性贫血等。本例患者除了表现出这些共同点外,还有一些特有表现如高钙血症、骨发育不良及慢性腹泻等,此外患者对T3的反应也说明仅为部分性T3抵抗。患者的这些特殊表现与突变的关联不是很明确,还需要进一步的研究或相同病例报道来获得证实。
发布于 15-06-12 19:23